Roothaan equations
The Roothaan equations are a representation of the Hartree-Fock equation in a non orthonormal basis set which can be of Gaussian-type or Slater-type. It applies to closed-shell molecules or atoms where all molecular orbitals or atomic orbitals, respectively, are doubly occupied. This is generally called Restricted Hartree-Fock theory.
The method was developed independently by Clemens C. J. Roothaan and George G. Hall in 1951, and is thus sometimes called the Roothaan-Hall equations.[1][2][3] The Roothaan equations can be written in a form resembling generalized eigenvalue problem, although they are not a standard eigenvalue problem because they are nonlinear:
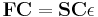
where F is the so-called Fock matrix (which depends on the coefficients C due to electron-electron interactions), C is a matrix of coefficients, S is the overlap matrix of the basis functions, and 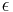 is the (diagonal, by convention) matrix of orbital energies. In the case of an orthonormalised basis set the overlap matrix, S, reduces to the identity matrix. These equations are essentially a special case of a Galerkin method applied to the Hartree-Fock equation using a particular basis set.
is the (diagonal, by convention) matrix of orbital energies. In the case of an orthonormalised basis set the overlap matrix, S, reduces to the identity matrix. These equations are essentially a special case of a Galerkin method applied to the Hartree-Fock equation using a particular basis set.
In contrast to the Hartree-Fock equations - which are integro-differential equations - the Roothaan-Hall equations have a matrix-from. Therefore they can be solved using standard techniques.
See also
References
- ^ Frank Jensen, Introduction to Computational Chemistry, John Wiley and Sons, 1999, pg 65 - 69, ISBN 0 471 98055
- ^ Roothaan, C. C. J. (1951). "New Developments in Molecular Orbital Theory". Reviews of Modern Physics 23: 69–89. Bibcode 1951RvMP...23...69R. doi:10.1103/RevModPhys.23.69.
- ^ Hall, G. G. (1951). "The Molecular Orbital Theory of Chemical Valency. VIII. A Method of Calculating Ionization Potentials". Proceedings of the Royal Society London A 205: 541–552. Bibcode 1951RSPSA.205..541H. doi:10.1098/rspa.1951.0048.